| Autore |
Discussione |
|
|
Luis 22
Utente


Citt�: Bari

755 Messaggi |
 Inserito il - 13 gennaio 2007 : 01:21:01 Inserito il - 13 gennaio 2007 : 01:21:01






|
Abbiamo avuto un interessante esercizio da svolgere a casa.
Partendo dalle masse di frammenti di una proteina incognita, ottenuti per spettrometria di massa, dobbiamo identificare la proteina usando le banche dati sia di profound che di mascot.
In particolare dobbiamo identificare 5 proteine e per ognuna di queste abbiamo 2 gruppi di masse dei frammenti, ottenute da 2 diversi operatori. Abbiamo saputo inoltre, che queste proteine sono fattori di trascrizione umani e che ci possono essere contaminazioni (abbiamo trovato in particolare cheratina).
Vi faccio un esempio, per un fattore di trascrizione che dobbiamo trovare abbiamo questi due gruppi di masse:
1)
945.5441
1036.4528
1118.5080
1179.5724
1235.5405
1307.6423
1365.6102
1383.6359
1475.7367
1499.6610
1515.7321
1516.6570
1547.7117
1638.8151
1707.7346
1790.8690
1940.8632
1954.0011
1960.8680
2082.9160
2311.9867
2383.8575
2)
946.3879
952.4364
965.3981
982.4604
1037.3976
1054.3623
1069.3311
1144.4008
1314.4281
1484.4701
1607.0594
1651.7399
1661.7347
Ora, usando profound, non si riescono ad inviduare in maniera precisa questi fattori di trascrizione. Per mascot, non ne parliamo!!!
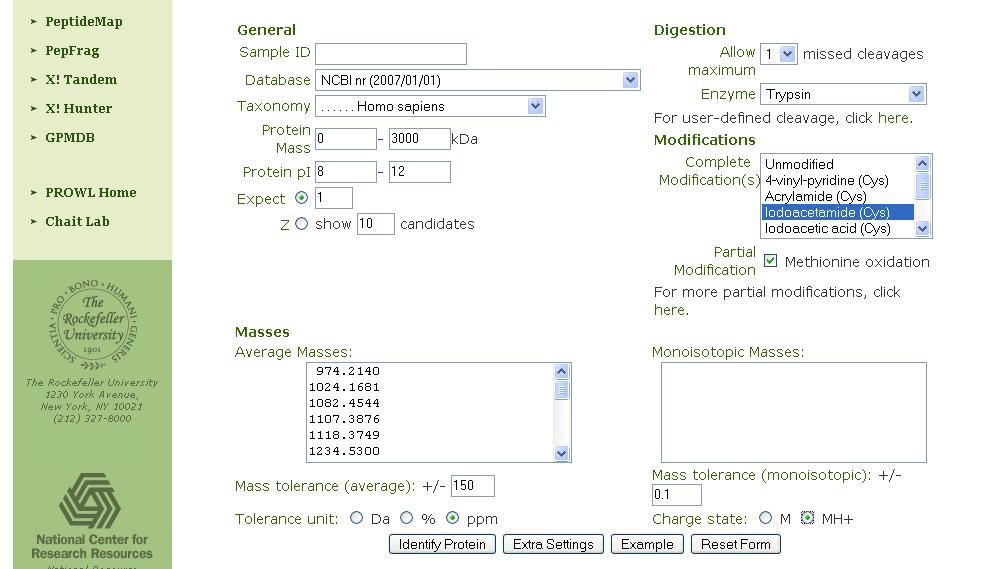
In allegato vi faccio vedere che parametri uso, in particolare:
- modificazioni con iodoacetammide
- parziali modificazioni (ossidazione)
- mass tolerance: 150 ppm
- MH+
- pI: 8-12
Per riuscire ad individuare i fattori di trascrizione bisogner� giocare sui vari parametri, anche in base alle caratteristiche dei fattori di trascrizione.
Ad esempio, io ho usato come pI il range 8-12, in quanto ho trovato che queste proteine hanno valori che cadono all'incirca in questo range.
Avete suggerimenti su come modificare altri parametri o conoscete altre caratteristiche dei fattori di trascrizione utili per la loro individuazione?
Immagine:
79,76 KB
|
 
La vita e i sogni sono fogli di uno stesso libro. Leggerli in ordine � vivere, sfogliarli a caso � sognare. (Arthur Schopenhauer)& |
|
|
|
|
domi84
Moderatore


Citt�: Glasgow
1724 Messaggi |
 Inserito il - 13 gennaio 2007 : 11:44:57 Inserito il - 13 gennaio 2007 : 11:44:57
|
Quando facevo io questi esercizi eravamo soliti dividere tutti i valori in gruppi di 6-7 che inserivamo nel programma...e questo si faceva svariate volte, finch� non usciva una proteina. Di solito scartavamo i picchi pi� "brutti" dallo spettro. Tu hai solo i valori o anche lo spettro?
Cmq mi spiego meglio:
Se inserisci tutti e 15 valori il programma cercher� una proteina che presenta tutti e 15 i valori,quindi avrai uno score molto basso perch� fra i valori che hai inserito ci sono molti valori che non appartengono alla prot che tu cerchi! Noi invece inserivamo solo 6-7 valori alla volta (casuali) per� svariate volte...secondo il prof che ci faceva le lezioni era pi� probabile individuare la prot in questo modo. |
Il mio blog: http://domi84.blogspot.com/
Le foto che ho scattato... |
 |
|
|
Luis 22
Utente
Citt�: Bari
755 Messaggi |
Inserito il - 13 gennaio 2007 : 11:57:52
|
Purtroppo abbiamo avuto solo i valori, niente spettro!
Comunque, ho capito il tuo ragionamento, in effetti per alcune proteine abbiamo trovato tra i risultati, la cheratina, evidente contaminante!
|
La vita e i sogni sono fogli di uno stesso libro. Leggerli in ordine � vivere, sfogliarli a caso � sognare. (Arthur Schopenhauer)& |
|
|
|
Luis 22
Utente
Citt�: Bari
755 Messaggi |
Inserito il - 13 gennaio 2007 : 12:33:20
|
| Ma facendo quindi pi� prove, poi � possibile combinare i dati ottenuti per vedere quello che � pi� comune? |
La vita e i sogni sono fogli di uno stesso libro. Leggerli in ordine � vivere, sfogliarli a caso � sognare. (Arthur Schopenhauer)& |
|
|
|
domi84
Moderatore
Citt�: Glasgow
1724 Messaggi |
Inserito il - 13 gennaio 2007 : 14:01:58
|
Citazione:
Messaggio inserito da Luis 22
Ma facendo quindi pi� prove, poi � possibile combinare i dati ottenuti per vedere quello che � pi� comune?
No, cos� otterresti un dato statistico. Io invece (usavo mascot) cliccavo sul nome della prot che credevo probabile, poi cliccavo su "show predicted peptides also", questo ti mostrer� tutti i peptidi che si formano di quella prot, con i rispettivi pesi.
Ora, se molti dei pesi di questi peptidi compaiono sul tuo spettro: la proteina � giusta!!!
Se invece nessuno di quei pesi compare sul tuo spettro:la proteina � sbagliata...ritenta, sarai pi� fortunato!!!
Io facevo cos�, e anche se il procedimento � molto manuale, lungo, e laborioso, mi trovavo bene!
Inoltre credo che anche i pi� esperti per fare il Fingerprinting ci mettono pi� di 10min!!! |
Il mio blog: http://domi84.blogspot.com/
Le foto che ho scattato... |
|
|
|
Luis 22
Utente
Citt�: Bari
755 Messaggi |
Inserito il - 28 gennaio 2007 : 20:32:47
|
Ok, grazie per l'aiuto! |
La vita e i sogni sono fogli di uno stesso libro. Leggerli in ordine � vivere, sfogliarli a caso � sognare. (Arthur Schopenhauer)& |
|
|
|
domi84
Moderatore
Citt�: Glasgow
1724 Messaggi |
|
|
Luis 22
Utente
Citt�: Bari
755 Messaggi |
Inserito il - 11 febbraio 2007 : 23:59:19
|
| Insomma, i campioni erano contaminati in una maniera pazzesca... |
La vita e i sogni sono fogli di uno stesso libro. Leggerli in ordine � vivere, sfogliarli a caso � sognare. (Arthur Schopenhauer)& |
|
|
|
LaP
Nuovo Arrivato

Citt�: Milano-Bologna
14 Messaggi |
Inserito il - 02 luglio 2008 : 23:31:15
|
in ritardo di pi� di un anno ma lascio comunque qualche commento.....
Non sono daccordo con l'approccio spiegato dal prof di domi84. Se in linea teorica il ragionamento � pi� che corretto, l'alterazione del dato inserito altera il dato in uscita che in questo modo non � pi� reale ma un artefatto manuale. In poche parole: chi e come valida l'identificazione da un processo di 15 cicli di identificazione prendendo le masse random? e poi come definire random la scelta arbitraria di un operatore umano?
In generale quando si fanno delle identificazione da digestione triptica si sa che sono presenti delle contaminazioni tipiche di ogni laboratorio/processo. Tra queste oltre alle cheratine (1, 9, 10 le pi� presenti) vi sono le varie BSA ed Ig (per chi fa IP), ecc.
Limitandoci solo alle cheratine, utilizzando profound (ora non pi� attivo, vi consiglio aldente) dopo la prima identificazione potete cercare i vostir contaminanti riconosciuti dal software (con i parametri di errore prestabiliti), validarli anche se questi hanno uno score basso e non significativo e risottomettere la lista cos� processata.
Mai, ripeto mai rimuovere le masse manualmente! la valutazione dell'operatore non � mai precisa come l'errore strumentale (pochi ppm). Solo il software considera tutti i parametri!
Buon lavoro |
|
|
| |
Discussione |
|



