| Autore |
Discussione |
|
|
carmilla983
Nuovo Arrivato
Prov.: al
Citt�: alessandria

59 Messaggi |
 Inserito il - 16 febbraio 2008 : 16:54:53 Inserito il - 16 febbraio 2008 : 16:54:53




|
avrei bisogno che qualcuno mi spiegasse in modo molto chiaro e preciso il microscopio a fluorescenza convenzionale (spt il cammino ottico che non riesco a capire perfettamente), pregi e difetti.
l'ideale sarebbe se qlc potessi aiutarmi nella comparazione di questo microscopio con quello confocale laser.
vi ringrazio

|
|
|
|
|
chick80
Moderatore


Citt�: Edinburgh
11491 Messaggi |
 Inserito il - 17 febbraio 2008 : 01:39:44 Inserito il - 17 febbraio 2008 : 01:39:44
|
Allora, il microscopio a epifluorescenza (che � quello generalmente usato in biologia) funziona secondo questo schema (preso da wikipedia):

In pratica hai una luce (il "sole" sulla destra) che solitamente � una lampada a xenon o mercurio, che generalmente emettono nel range UV/visibile. La lampada emette luce in un range molto ampio di luce, ma tu vuoi eccitare il fluoroforo con una determinata lunghezza d'onda. Per questo scopo usi un filtro di eccitazione che fa passare solo una certa lunghezza d'onda o, per essere pi� precisi, fa comunque passare un range di lunghezze d'onda, ma molto pi� ristretto di quello emesso dalla lampada. Ad es. potrebbe far passare la luce fra 410 e 430 nm e se il tuo fluoroforo viene eccitato a 420 sei a posto! Nel disegno il filtro fa passare solo la luce blu.
A questo punto la luce arriva ad uno specchio dicroico. Questi sono specchi particolari che possono riflettere la luce o farla passare a seconda della lunghezza d'onda. Nell'esempio, lo specchio dicroico riflette la luce blu, che quindi cambia direzione e va gi� verso il campione attraverso l'obiettivo.
Il campione viene quindi eccitato e, nell'esempio, emette luce verde che passa nuovamente attraverso l'obiettivo ed arriva allo specchio dicroico. Lo specchio questa volta fa passare la luce verde che pu� quindi arrivare al detector (es. una telecamera oppure semplicemente l'occhio dell'osservatore!)
Generalmente prima del detector c'� un altro filtro, detto filtro di emissione, che ti fa vedere solo una parte delle lunghezze d'onda emesse e questo � utile per eliminare eventuale altra luce (generata per qualsiasi motivo) che sia passata nell'obiettivo.
Il grosso vantaggio di questo sistema � che � relativamente semplice da usare e poco costoso (rispetto alle alternative) ed ha una medio-buona sensibilit�. Uno degli svantaggi � che quando vai ad illuminare il tuo campione lo illumini completamente, quindi TUTTE le molecole di fluoroforo nel campione sono eccitate, anche quelle che non stai effettivamente osservando (es quelle fuori dal piano focale). Questo porta ad un fenomeno noto come "bleaching" (bleach vuol dire candeggina, quindi si potrebbe tradurre "sbiancamento") che consiste nella perdita di fluorescenza del campione dopo che sta troppo tempo sotto la luce del microscopio. Anche per questo i preparati fluorescenti vanno generalmente tenuti al buio. Se poi il tuo campione � vivo (es. una fettina di tessuto fresco) si possono avere fenomeni di fototossicit� per cui le cellule vengono danneggiate da una continua esposizione alla radiazione eccitante, es. per formazione di radicali liberi durante il processo di eccitazione/emissione dei fluorofori.
L'altro grosso problema � la luce che viene dagli altri piani focali. Poich� stai eccitando tutto il tuo campione, la luce osservata (quella verde che arriva al detector) non arriva necessariamente tutta dal piano focale che stai osservando, ma pu� arrivare anche da pi� sopra o da pi� sotto, e questo pu� risultare in immagini poco nitide. Inoltre se stai cercando di fare una colocalizzazione di due molecole marcate con fluorofori diversi non puoi essere certo che i due colori siano esattamente colocalizzati, perch� potrebbero essere su due piani diversi.
Alcuni di questi problemi possono essere risolti o perlomeno ridotti dall'uso di un confocale.
----
Il confocale funziona in maniera simile al microscopio a fluorescenza:

presa da http://www.microscopyu.com
Innanzitutto la luce viene generata da un laser. I laser usati per la microscopia confocale emettono una lunghezza d'onda fissa quindi non hai bisogno di filtri di eccitazione. Hai ancora una volta il tuo specchio dicroico che fa passare la luce, nel disegno verde, e l'obiettivo che la focalizza sul campione. Il campione viene nuovamente eccitato ed emette luce (in rosso) sul piano focale e da altri piani. E qui arriva il trucco! La luce torna allo specchio dicroico, viene fatta passare e poi passa da un "pinhole", un buchino molto piccolo la cui dimensione pu� essere regolata dall'osservatore che fa passare SOLO la luce dal piano focale. (devi seguire un po' le frecce nel disegno).
A questo punto arriva al detector che nel caso del confocale � generalmente un fotomoltiplicatore (PMT) che serve per amplificare il segnale, in quanto gli arriva molta meno luce che in un microscopio convenzionale.
Vantaggi del confocale:
1) il laser viene focalizzato dall'obiettivo sul campione, quindi arriva in un solo punto del campione. Quindi, nonostante tu abbia comunque bleaching sull'asse z, cio� nella profondit� del campione, non avrai bleaching nelle zone che non stai guardando. D'altra parte la luce del laser � generalmente pi� forte (anche se pu� essere modulata) quindi nella zona dove stai osservando il bleaching � molto pi� forte.
2) vedi solo la luce del piano focale, quindi hai immagini molto nitide e pulite, dove vedi solo il piano focale e niente altro.
3) se prendi immagini da diversi piani focali puoi fare una ricostruzione in 3D del campione che fa sempre scena su una pubblicazione!!! 
4) siccome puoi focalizzare il laser solo su di un punto del campione puoi fare esperimenti molto eleganti di fotoattivazione, in cui hai un composto fotoattivabile che puoi attivare solo in una certa ristretta regione del tuo campione. Questo pu� essere ad esempio un fluoroforo che normalmente non emette, ma una volta che viene colpito da una particolare lunghezza d'onda si attiva e comincia a fluorescere oppure un composto chimico "caged" cio� messo in una gabbia fotosensibile. Il composto � inattivo fino a che la gabbia non viene distrutta dal laser e rilascia il composto solo in una ristretta regione del tuo campione.
Il grosso svantaggio � che il confocale costa di pi�, � pi� complesso da mantenere (di solito ti serve avere un tecnico che se ne prende cura) ed � uno strumento molto pi� delicato.
----
Ulteriori miglioramenti nell'acquisizione delle immagini si possono ottenere utilizzando il microscopio a due fotoni che usa un principio differente (ed abbastanza complesso) che ha tra le altre cose il vantaggio di permetterti di focalizzare su regioni pi� in profondit� nel campione e di generare pochissimo bleaching. |
Sei un nuovo arrivato?
Leggi il regolamento del forum e presentati qui
My photo portfolio (now on G+!) |
 |
|
|
carmilla983
Nuovo Arrivato
Prov.: al
Citt�: alessandria
59 Messaggi |
Inserito il - 17 febbraio 2008 : 09:44:35
|
ti ringrazio davvero per la chiarezza e precisione!!!adesso si che ho capito...il microscopio a due fotoni sarebbe quello che viene anche chiamato multifotone? e qst come funziona? perch� ho seguito un corso di tecniche fisiche x la biologia ma ho capito ben poco nonostante gli appunti e dispense.
|
|
|
|
carmilla983
Nuovo Arrivato
Prov.: al
Citt�: alessandria
59 Messaggi |
Inserito il - 17 febbraio 2008 : 11:38:59
|
ehm...� sorto un altro problema..la generazione dell'immagine da un microscopio confocale. premettendo che il laser analizza la fetta punto per punto, la ricostruzione dell'immagine come avviene? da che cosa pu� essere influenzata?
nelle lezioni, il prof parla di tempi degli specchi galvanometrici e della necessit� di sincronizzare..che cosa sono qst specchi?e cosa s deve sincronizzare per ottenere una migliore immagine del preparato?
grazie a chi mi aiuta! |
|
|
|
chick80
Moderatore
Citt�: Edinburgh
11491 Messaggi |
Inserito il - 17 febbraio 2008 : 13:23:59
|
Allora, partiamo dagli specchi galvanometrici: come ti dicevo il laser colpisce un solo punto del tuo campione. Tu per� di solito vorrai avere un'immagine un po' pi� grande di un solo punto...
Si usano quindi questi specchi galvanometrici che sono specchi che possono ruotare e riflettere il laser in modo che vada in un certo specifico punto. Se fai muovere lo specchio in modo opportuno puoi prendere mano a mano l'immagine di tutti i punti nel rettangolo che ti interessa.
Ad es, se questo � il tuo campione (con O indicata l'area che vuoi analizzare):
-----------------
| |
| OOOOO |
| OOOOO |
| |
-----------------
Cominci e illumini il primo punto dell'area di interesse, poi il successivo, poi il successivo e cos� via
-----------------
| |
| xOOOO |
| OOOOO |
| |
-----------------
-----------------
| |
| OxOOO |
| OOOOO |
| |
-----------------
-----------------
| |
| OOxOO |
| OOOOO |
| |
-----------------
etc etc
Mettendo insieme le immagini ottenute illuminando ciascun punto otterrai quindi l'immagine finale.
Il problema � che, soprattutto se il campione � costituito da cellule vive, quindi in costante cambiamento, il microscopio deve essere abbastanza veloce a passarsi tutti i punti. Questo dipende da molti fattori ma sicuramente i pi� importanti sono: 1) la bont� dell'apparecchiatura 2) la fluorescenza del campione. Se la fluorescenza � molto forte baster� un tempo di esposizione molto piccolo per ogni punto 3) la grandezza dell'area che stai guardando.
Quando il tuo prof parla di sincronizzazione intende proprio questo. Tu in teoria vuoi che le immagini di tutti i punti della tua area siano sincronizzate, cio� prese allo stesso istante (cosa che succede ad es in un microscopio ad epifluorescenza, questo � un vantaggio che ha rispetto al confocale), anche se ci� � tecnicamente impossibile perch� comunque passer� del tempo, anche se minimo, fra l'immagine di un punto e quella del successivo.
-------
Per quanto riguarda l'eccitazione multifotoni. In pratica perlomeno per adesso si parla solo di eccitazione a 2 fotoni, quella a pi� di 2 fotoni � stata ipotizzata e credo in parte sperimentata ma ci sono problemi tecnici per poterla effettivamente utilizzare a fini pratici.
Immagine (da http://www.microscopyu.com/articles/fluorescence/multiphoton/multiphotonintro.html )

In pratica nel microscopio a fluorescenza convenzionale (epifluorescenza o confocale) tu hai un'eccitazione ad 1 fotone (1PE): un fotone colpisce il fluoroforo, questo arriva ad un livello quantico eccitato e torna al livello di base emettendo fluorescenza.
Nell'eccitazione a 2 fotoni (2PE) colpisci il fluoroforo con luce dalla lunghezza d'onda pi� lunga (di solito si sta tra i 900 e i 1200 nm) che non ha energia sufficiente a farlo arrivare al livello superiore, ma lo fa andare in uno stato "virtuale" fra i due livelli. Se in un tempo molto breve (si parla di femtosecondi) un altro fotone a bassa energia colpisce il fluoroforo questo passa allo stato superiore, altrimenti ricade allo stato base senza emettere fluorescenza.
Ora, con l'utilizzo di speciali laser (es laser titanio-zaffiro) che emettono impulsi laser di breve durata si riesce a far s� che il laser ecciti le molecole di fluoroforo solo in un punto e solo nel piano di fuoco. Questo perch� anche se le molecole di piani fuori fuoco possono essere colpite da un primo fotone, la probabilit� che vengano colpite da un secondo fotone � praticamente nulla, perch� la densit� di fotoni al di fuori del piano focale � troppo bassa perch� ci� accada.
In questo caso non ti serve nemmeno pi� il pinhole, perch� TUTTA la luce che torna nell'obiettivo verr� dal punto che stiamo eccitando (anche il microscopio a 2 fotoni crea l'immagine punto per punto come il confocale), ma solo dal piano focale.
Altri vantaggi sono:
- meno fotodanneggiamento e bleaching, a parte nel punto che stiamo osservando.
- si possono ottenere immagini da parti pi� profonde del campione, in quanto la luce a lunghezze d'onda maggiori penetra pi� a fondo nel tessuto (in generale si pu� arrivare senza troppi problemi a 1mm di profondit�)
questo rende il microscopio a 2 fotoni ottimo per l'utilizzo per microscopia in vivo |
Sei un nuovo arrivato?
Leggi il regolamento del forum e presentati qui
My photo portfolio (now on G+!) |
|
|
|
carmilla983
Nuovo Arrivato
Prov.: al
Citt�: alessandria
59 Messaggi |
Inserito il - 18 febbraio 2008 : 12:03:03
|
sei davvero gentilissimo, grazie per avermi risposto cos� bene!
ehm..un quesito forse stupido..dunque adesso sto ascoltando una registrazione di un laboratorio che non ho potuto seguire sullo stesso argomento. alle persone presenti sta facendo vedere un microscopio a fluorescenza a campo largo, con (??)nomarsky e confocale. ma pu� essere lo stesso microscopio con le 3 funzioni insieme?
inoltre fa vedere un microscopio a fluorescenza invertito..ma deve essere sempre cos�? e che differenza c'� tra microscopio a fluorescenza normale e a campo largo?
ps:dove posso trovare immagini di queste tre funzioni e di un microscopio come quello di cui parla il prof tanto per farmi un'idea su quello di cui parla?
grazie10001000 |
|
|
|
chick80
Moderatore
Citt�: Edinburgh
11491 Messaggi |
Inserito il - 18 febbraio 2008 : 23:36:29
|
Citazione:
alle persone presenti sta facendo vedere un microscopio a fluorescenza a campo largo, con (??)nomarsky e confocale. ma pu� essere lo stesso microscopio con le 3 funzioni insieme?
S�! normalmente il microscopio confocale ha anche associato una lampada al mercurio o allo xenon per usarlo come normale microscopio a epifluorescenza, in modo che puoi guardare velocemente il tuo campione attraverso gli oculari del microscopio, spostarlo in modo da centrarlo su quello che vuoi vedere etc.
Nello stesso microscopio c'� anche una normale lampada ad incandescenza per vedere il tuo campione con luce trasmessa.
Il Normansky � un tipo di microscopia DIC (differential interference contrast) che in pratica sfrutta luce polarizzata per darti un'immagine pi� "3D" del tuo campione.
Vedi
http://en.wikipedia.org/wiki/Differential_interference_contrast_microscopy
Puoi facilmente andare da una funzione all'altra muovendo dei filtri a mano o in remoto (via computer).
Citazione:
inoltre fa vedere un microscopio a fluorescenza invertito..ma deve essere sempre cos�?
No, la luce pu� arrivare sia dall'alto (epifluorescenza, come ti ho spiegato sopra) che dal basso (microscopio invertito, meno comune per la fluorescenza).
Citazione:
che differenza c'� tra microscopio a fluorescenza normale e a campo largo
Microscopio a fluorescenza � il termine generale per tutti i tipi di microscopio che ti ho indicato nelle risposte precedenti. Wide field (campo largo) indica che tutto il campione � illuminato dalla luce e non un solo punto alla volta come nel confocale o nel 2 fotoni.
Citazione:
dove posso trovare immagini di queste tre funzioni e di un microscopio come quello di cui parla il prof tanto per farmi un'idea su quello di cui parla
Gooooooogle!
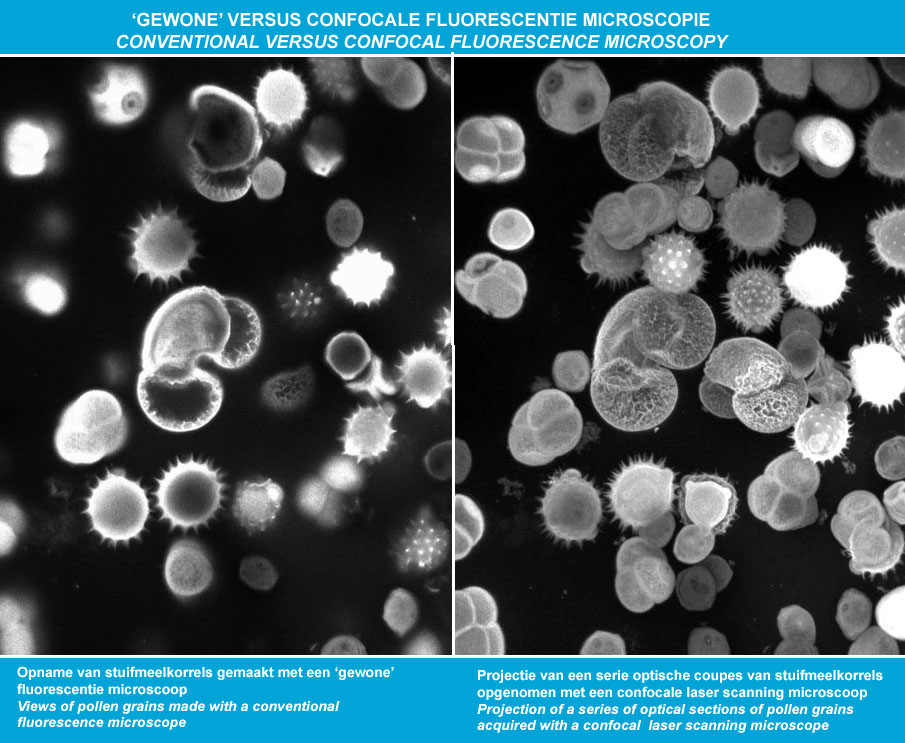
Ad es:
widefield (sx) vs. confocale (dx)
normansky (sopra) vs. luce trasmessa (sotto)

|
Sei un nuovo arrivato?
Leggi il regolamento del forum e presentati qui
My photo portfolio (now on G+!) |
|
|
|
carmilla983
Nuovo Arrivato
Prov.: al
Citt�: alessandria
59 Messaggi |
Inserito il - 19 febbraio 2008 : 09:55:11
|
intendevo,scusami, foto dei microscopi...cmq ne ho trovate. grazie, sei stato di grande aiuto!
il confocale normalmente � invertito, giusto?
ho aperto un altro topic x altre info per� provo a chiederti qui se potresti spiegarmi la differenza tra un microscopio a luce riflessa e trasmessa..il dic � a luce riflessa o trasmessa?ho visto delle immagini sul web ma non riesco a capire che propriet� sfruttano del campione qst due micro.
nel caso del confocale o del microscopio x fluorescenza, ha senso parlare di luce riflessa o trasmessa?
ultima cosa..nel microscopio a fluorescenza invertito, lui ha parlato di monocromatore, specchi e finestrella dove convogliare il fascio di luce..parla di diversi specchi nel micro x fluorescenza..ma non c'� ne uno solo, lo specchio dicroico??n ci capisco pi� nulla..dice tutto e il contrario di tutto...
ha detto che il fascio d luce esce da dietro ed attraverso una fibra ottica entra nel microscopio..perch� fa tuto questo percorso la luce?
grasie e scusami per le 1000 doamnde!
|
|
|
|
carmilla983
Nuovo Arrivato
Prov.: al
Citt�: alessandria
59 Messaggi |
Inserito il - 19 febbraio 2008 : 10:32:53
|
una delucidazione..1)filtro di eccitazione|emissione = monocromatore? nel micro a fluorescenza...
2) lampade ad arco con xenon o mercurio sono le sorgenti d eccitazione usate nei micro a fluorescenza? |
|
|
|
chick80
Moderatore
Citt�: Edinburgh
11491 Messaggi |
Inserito il - 19 febbraio 2008 : 23:05:51
|
Citazione:
il confocale normalmente � invertito, giusto?
Mah, noi ne abbiamo 2 invertiti e 2 con l'obiettivo sopra (mi sfugge il nome in italiano...). Dipende da che obiettivi usi: obiettivi ad immersione (in acqua o olio) devono per forza essere sopra al campione (altrimenti l'acqua o l'olio cascano gi�!), se vuoi un confocale per guardare cellule ad es. direttamente in una Petri puoi invece usare un invertito. Dipende essenzialmente dall'utilizzo che ne vuoi fare.
Citazione:
ho aperto un altro topic x altre info per� provo a chiederti qui se potresti spiegarmi la differenza tra un microscopio a luce riflessa e trasmessa..il dic � a luce riflessa o trasmessa
a questa domanda ti rispondo pi� tardi che sono un po' preso adesso 
Citazione:
Nel caso del confocale o del microscopio x fluorescenza, ha senso parlare di luce riflessa o trasmessa?
Non nel caso della fluorescenza, anche se, come dicevo sopra di solito questi microscopi hanno entrambe le funzioni (fluorescenza + luce visibile)
Citazione:
ultima cosa..nel microscopio a fluorescenza invertito, lui ha parlato di monocromatore, specchi e finestrella dove convogliare il fascio di luce..parla di diversi specchi nel micro x fluorescenza..ma non c'� ne uno solo, lo specchio dicroico
Beh, lo schema che ti ho fatto io era abbastanza semplificato, in realt� ci sono vari filtri che devi aggiungere pi� che altro per problemi tecnici.
Citazione:
ha detto che il fascio d luce esce da dietro ed attraverso una fibra ottica entra nel microscopio..perch� fa tuto questo percorso la luce?
Conta che la lampada per la fluorescenza non � solo una lampadina che avviti dietro al microscopio! Di solito � contenuta in una "scatola" piuttosto ingombrante che devi collegare in qualche modo al microscopio... e questo qualche modo � una fibra ottica.
Citazione:
filtro di eccitazione|emissione = monocromatore? nel micro a fluorescenza...
Non esattamente, anche se fanno pi� o meno lo stesso lavoro. Un monocromatore (vedi: http://it.wikipedia.org/wiki/Monocromatore ) � basato su un reticolo di diffrazione o su di un prisma, mentre un filtro � semplicemente un disco di vetro particolare tipo quelli che metti sulle macchine fotografiche.
Citazione:
lampade ad arco con xenon o mercurio sono le sorgenti d eccitazione usate nei micro a fluorescenza?
S�, ma non per il confocale o per il due fotoni che invece usano laser (ad es. laser ad argon o a Ti:zaffiro) |
Sei un nuovo arrivato?
Leggi il regolamento del forum e presentati qui
My photo portfolio (now on G+!) |
|
|
|
carmilla983
Nuovo Arrivato
Prov.: al
Citt�: alessandria
59 Messaggi |
Inserito il - 20 febbraio 2008 : 10:41:01
|
grazie mille ancora...
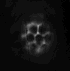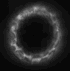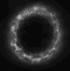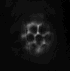
dunque avrei trovato questa immagine nella mia dispensa
http://img214.imageshack.us/my.php?image=immagineda9.jpg
ma non riesco a capire perch� nel caso della fluorescenza a campo largo il pattern di illuminazione � grande ma la psf ha quella forma..lui ha farfugliato qlcosa sul fatto che l'illuminazione tende a decrescere ed espandersi ma non � simmetrica, � distorta..eh??
potresti mica spiegarmi perch� vengono cos� le PSF e i pattern di illuminazione sono schematizzati cos� nel caso del confocale e multifotone?
per il microscopio a luce trasmessa e riflessa, m sono aggiustata..grazie cmq!!! |
|
|
|
chick80
Moderatore
Citt�: Edinburgh
11491 Messaggi |
Inserito il - 20 febbraio 2008 : 12:54:16
|
La conosco quell'immagine! E' presa da:
To 5D and beyond: quantitative fluorescence microscopy in the postgenomic era.
Comunque sia: i pattern di illuminazione sono cos� perch� nel confocale tu hai comunque luce che arriva da piani fuori fuoco in quanto molecole di fluoroforo in quei piani possono essere colpite dal laser ed essere eccitate. Nell'immagine finale non vedi la luce da questi piani perch� il pinhole seleziona solo il piano focale (o comunque una ristretta serie di piani focali, dipende da quanto lo apri).
Nel 2 fotoni, invece, poich� solo nel punto di fuoco la densit� di fotoni � abbastanza alta da avere eccitazione a due fotoni, l'illuminazione dei piani fuori fuoco � praticamente nulla.
Per quanto riguarda la PSF. Nel wide field se hai un oggetto puntiforme vedrai la sua immagine nitida nel piano focale e sempre pi� sfocata e diffusa man mano che te ne allontani. E' proprio per questo che hai una PSF (point spread function) a forma di doppio cono. Il punto centrale in cui lo spread � minimo � sul piano focale.
Nel confocale e nel 2 fotoni ci� non avviene perch� non hai (o hai solo in minima parte) luce derivante da piani fuori fuoco.
Non chiedermi perch� da una parte usa il verde e dall'altra il rosso per disegnare la PSF... non saprei proprio dirti!
PS: scusa la curiosit�... ma che corso di laurea stai seguendo? A noi non ce le facevano studiare tutte 'ste cose!
|
Sei un nuovo arrivato?
Leggi il regolamento del forum e presentati qui
My photo portfolio (now on G+!) |
|
|
|
carmilla983
Nuovo Arrivato
Prov.: al
Citt�: alessandria
59 Messaggi |
Inserito il - 20 febbraio 2008 : 13:00:20
|
sono al secondo anno d specialistica di biologia.il corso � tecniche fisiche per la biologia..no comment sulla quantit� di cose relative alla fisica che ha cercato di spiegare. ho davvero difficolt� a capire certi concetti in microscopia visto che l'abbiamo fatta solo al primo anno e superficialmente. in questo caso si va moooolto in profondit�..non ho idea d cosa poi chieda...
scusami se ho intasato il forum con mille domande...ma posso chiederti qualche aiuto se ho ancora qualche dubbio?
grazie..m sei davvero di aiuto!
 |
|
|
|
chick80
Moderatore
Citt�: Edinburgh
11491 Messaggi |
Inserito il - 20 febbraio 2008 : 13:13:01
|
Chiedi pure, non ti preoccupare! Sono sicuro che tutte queste informazioni potranno essere preziose anche per qualcun altro!
 |
Sei un nuovo arrivato?
Leggi il regolamento del forum e presentati qui
My photo portfolio (now on G+!) |
|
|
|
carmilla983
Nuovo Arrivato
Prov.: al
Citt�: alessandria
59 Messaggi |
Inserito il - 21 febbraio 2008 : 10:27:57
|
allora ne approfitto per chiedere una breve spiegazione della microscopia denominata STED
..ho trovato la traduzione di stimulated emission-depletion microscopy sia come micro a deplezione mediante emissione stimolata sia come ad emissione-deplezione stimolata..ma quale � quella giusta?
grasie!
nb:ho notato che le lunghezze d'onda dei laser usati nel confocale sono approssimativamente tra i 325 nm di elio-cadmio a 647 nm di cripton-argon..come mai n sfiorano il campo degli uv?
anche per confocale uso sonde fluorescenti vero?
gracias |
|
|
|
chick80
Moderatore
Citt�: Edinburgh
11491 Messaggi |
Inserito il - 21 febbraio 2008 : 12:01:14
|
Citazione:
..ho trovato la traduzione di stimulated emission-depletion microscopy sia come micro a deplezione mediante emissione stimolata sia come ad emissione-deplezione stimolata..ma quale � quella giusta?
Non conosco la tecnica sinceramente, ma come lo tradurrei come microscopia a delplezione emissione-stimulata (che nomi!)
Citazione:
nb:ho notato che le lunghezze d'onda dei laser usati nel confocale sono approssimativamente tra i 325 nm di elio-cadmio a 647 nm di cripton-argon..come mai n sfiorano il campo degli uv?
Esistono anche laser nell'UV, non so se ci siano pi� problemi tecnici nel costruirli (e quindi si usano meno)
Citazione:
anche per confocale uso sonde fluorescenti vero?
Certo! tutti i microscopi di cui si parlava sopra usano sonde fluorescenti. |
Sei un nuovo arrivato?
Leggi il regolamento del forum e presentati qui
My photo portfolio (now on G+!) |
|
|
|
carmilla983
Nuovo Arrivato
Prov.: al
Citt�: alessandria
59 Messaggi |
Inserito il - 21 febbraio 2008 : 12:19:56
|
http://www.mpibpc.gwdg.de/groups/hell/STED.htm
trovato ma n ci capisco molto...dunque ci sono due impulsi laser. il primo si riflette in una emissione per fluorescenza e l'altro? riporto "The excitation pulse is immediately followed by a depletion pulse, dubbed 'STED-pulse'. The STED pulse is red-shifted in frequency to the emission spectrum of the dye, so that its lower energy photons act ideally only on the excited dye molecules, quenching them to the ground state by stimulated emission. The net effect of the STED pulse is that the affected excited molecules cannot fluoresce because their energy is dumped and lost in the STED pulse. By spatially arranging the STED pulse in a doughnut mode, only the molecules at the periphery of the spot are ideally quenched. In the center of the doughnut, where the STED pulse is vanishing, fluorescence ideally remains unaffected"
avviene il quenching (sbiancamento)della periferia mentre il centro non viene colpito da questo fenomeno... il quenching � paragonabile al bleaching? il quenching � paragonabile al bleaching? |
|
|
|
carmilla983
Nuovo Arrivato
Prov.: al
Citt�: alessandria
59 Messaggi |
Inserito il - 21 febbraio 2008 : 19:03:20
|
un altro chiarimento urgente..dunque le aberrazioni possono essere cromatiche e monocromatiche. le prime s suddividono in longitudinali e laterali. ho cercato nella rete..tante spiegazioni ma nessuna terra terra che possa capire io...che differenza c'� tra longitudinali e laterali?
per le monocromatiche abbiamo coma, astigmatismo dei fasci obliqui, aberr.sferica, distorsione e curvatura di campo..le prime non le ho proprio capite. potreste aiutarmi?
ma le ultime tre non sono un po'tutte la stessa cosa?
grazie davvero..ho stampato tnt cose in italiano e in inglese ma sono venute fuori poche idee e confuse..aiutatemi per favore! grazie grazie |
|
|
|
carmilla983
Nuovo Arrivato
Prov.: al
Citt�: alessandria
59 Messaggi |
Inserito il - 21 febbraio 2008 : 21:00:41
|
| riguardo all'immagine del pattern di illuminazione e della psf nel caso del confocale, viene rappresentata come ovalizzata invece dovrebbe essere sferica. il motivo secondo il prof � che in realt� n essendo il microscopio simmetrico, la psf viene allungata..come, il microscopio non � simmetrico? |
|
|
|
chick80
Moderatore
Citt�: Edinburgh
11491 Messaggi |
Inserito il - 22 febbraio 2008 : 07:35:06
|
Citazione:
avviene il quenching (sbiancamento)della periferia mentre il centro non viene colpito da questo fenomeno...il quenching � paragonabile al bleaching?
No! Il quenching (smorzamento) � un fenomeno diverso dal bleaching.
Hai bleaching quando continui ad eccitare per lungo tempo un fluoroforo. Piano piano il fluoroforo perde fluorescenza e dopo un tot non � pi� fluorescente (a volte lasciarlo per un po' al buio aiuta).
Il quenching (o smorzamento) � un fenomeno che si ottiene quando usi un qualche sistema per smorzare la fluorescenza di un fluoroforo. Ad esempio si ha quando hai il fluoroforo vicino ad un altro gruppo (chiamato quencher) che ne assorbe la luce emessa. Se il quencher � abbastanza vicino al fluoroforo, la luce emessa dal fluoroforo viene assorbita dal quencher e quindi non viene rilevata dall'osservatore. Se il quencher � lontano dal fluoroforo, invece, rilevi la luce.
Nel caso particolare della STED, invece, il quenching viene indotto con un fenomeno chiamato "emissione stimolata" (vedi: http://en.wikipedia.org/wiki/Stimulated_emission ) per cui una molecola nel suo stato eccitato se colpita da un fotone torna al suo stato base e genera un altro fotone identico a quello da cui � stata colpita.
La tecnica consiste nell'illuminare il tuo campione con 2 laser sincronizzati. Uno eccita la molecola, mentre l'altro, a lunghezza d'onda pi� alta (quindi spostato verso il rosso), viene usato per fare "quenching" dell'eccitazione generata dal primo laser. Il trucco sta nel focalizzare questo laser quencher in un'area a forma di "ciambella" attorno a quello che eccita, in questo modo in pratica ottieni la fluorescenza solo dal punto centrale ed aumenti la risoluzione. Questa tecnica ti permette di superare il limite di diffrazione, e quindi di vedere distintamente punti pi� vicini della minima distanza teoricamente ottenibile con quel laser e obiettivo.
Citazione:
he differenza c'� tra longitudinali e laterali
Le aberrazioni longitudinali si hanno quando metti a fuoco diverse lunghezze d'onda a diversa distanza dall'obiettivo (quindi su diversi piani focali), mentre quelle laterali quando metti a fuoco diverse lunghezze d'onda in differenti parti del piano focale.
E' la tipica cosa che vedi in quando usi una lente scadente per la macchina fotografica e vedi qualcosa tipo:

Come vedi il rosso (a dx) e l'azzurro (a sx) non sono messi a fuoco correttamente sul piano focale dell'obiettivo.
Citazione:
coma, astigmatismo dei fasci obliqui, aberr.sferica, distorsione e curvatura di campo
Coma: dipende da imperfezioni della lente o dal fatto che il microscopio non sia correttamente allineato. In pratica luce derivante da punti lontani dall'asse della lente vengono distorti (perch� messi a fuoco su un piano focale diverso).
Ad es:

astigmatismo: la lente ha piani focali diversi per piani perpendicolari (es. diverso per orizzontale e verticale). Hai a fuoco le linee orizzontali ma non le verticali o viceversa. Noi astigmatici conosciamo bene gli effetti di questa aberrazione!

aberrazione sferica

Sopra hai una lente ideale, che mette a fuoco tutti i raggi in uno stesso punto.
In pratica, � difficile costruire una lente del genere e ti trovi in una situazione tipo quella di sotto (molto esagerata nel disegno): in pratica i raggi che arrivano dai bordi della lente vengono focalizzati in un piano pi� vicino di quelli che passano dal centro.
Ottieni quindi una curvatura di campo perch� la lente mette a fuoco i punti su di una superficie curva, non sul piano focale.
Vedi qualcosa tipo:

Come vedi il centro � sul piano focale mentre attorno hai tutto sfocato.
In questa immagine vedi anche dell'astigmatismo specialmente sulle croci 2 e 3
In questo caso hai quindi distorsione dell'immagine. Nell'esempio hai una distorsione a "pincushion" (puntaspilli), se il piano fosse curvo nell'altra direzione (entrando nel monitor) avresti una "barrel distorsion", cio� distorsione a "barile".
L'immagine viene
Qui trovi esempi interattivi di tutte queste aberrazioni:
http://micro.magnet.fsu.edu/primer/lightandcolor/opticalaberrations.html
Citazione:
riguardo all'immagine del pattern di illuminazione e della psf nel caso del confocale, viene rappresentata come ovalizzata invece dovrebbe essere sferica. il motivo secondo il prof � che in realt� n essendo il microscopio simmetrico, la psf viene allungata..come, il microscopio non � simmetrico?
Di questo proprio non ho idea sorry... |
Sei un nuovo arrivato?
Leggi il regolamento del forum e presentati qui
My photo portfolio (now on G+!) |
|
|
|
carmilla983
Nuovo Arrivato
Prov.: al
Citt�: alessandria
59 Messaggi |
Inserito il - 22 febbraio 2008 : 09:49:02
|
sto cercando di capire le aberrazioni leggendo le spiegazioni da quel sito ma non capisco lo stesso..sono proprio una frana in fisica
ehm non ho mica capito le aberrazioni longitudinali e laterali. cosa intendi con "a fuoco diverse lunghezze d'onda a diversa distanza dall'obiettivo" e "a fuoco diverse lunghezze d'onda in differenti parti del piano focale"?che differenza c'�?
scusami se ti faccio ripetere i concetti .. |
|
|
|
chick80
Moderatore
Citt�: Edinburgh
11491 Messaggi |
Inserito il - 22 febbraio 2008 : 10:34:33
|
fuoco diverse lunghezze d'onda a diversa distanza dall'obiettivo = ad es. il rosso � a fuoco a 1 mm di distanza, il verde a 1.5 mm e il blu a 2 mm
a fuoco diverse lunghezze d'onda in differenti parti del piano focale = es. a sx della lente c'� a fuoco il rosso ma non il verde, a dx c'� a fuoco il verde ma non il rosso etc. |
Sei un nuovo arrivato?
Leggi il regolamento del forum e presentati qui
My photo portfolio (now on G+!) |
|
|
|
carmilla983
Nuovo Arrivato
Prov.: al
Citt�: alessandria
59 Messaggi |
Inserito il - 22 febbraio 2008 : 12:01:08
|
capito...
per quanto riguarda il quenching, esso assorbe l'energia emessa da un fluoroforo e pu� esso stesso riemettere?
in poche parole, vengono usati i quenchers nella tecnica FRET?
altra domanda sempre sul confocale...la unit scanning � composta da vari elementi a guardando qst immagine non riesco a capire che percorso faccia il laser..mi hai detto che nel caso del confocale non abbiamo bisogno di un filtro di eccitazione eppure compare nell'image cos� come quello di emissione..perch�?
http://img301.imageshack.us/my.php?image=unitscanningqf9.jpg
ho letto la spiegazione del sito da cui hanno preso questa immagine ma il cammino ottico � spiegato in maniera confusa secondo me..chiedo a te se puoi illuminarmi (magari n con il laser!)
aiuto..non ho capito l'astigmatismo dei fasci obliqui...con termini semplici avrei bisogno di qualche info in pi�.
grasie |
|
|
|
chick80
Moderatore
Citt�: Edinburgh
11491 Messaggi |
Inserito il - 23 febbraio 2008 : 23:21:44
|
Citazione:
per quanto riguarda il quenching, esso assorbe l'energia emessa da un fluoroforo e pu� esso stesso riemettere?
in poche parole, vengono usati i quenchers nella tecnica FRET?
Non ricordo se esista un termine diverso per i quenchers che poi riemettono a lunghezza d'onda diversa (almeno, non mi viene in mente), per� il fenomeno � praticamente lo stesso.
Citazione:
altra domanda sempre sul confocale...la unit scanning � composta da vari elementi a guardando qst immagine non riesco a capire che percorso faccia il laser..mi hai detto che nel caso del confocale non abbiamo bisogno di un filtro di eccitazione eppure compare nell'image cos� come quello di emissione..perch�?
Ho sempre pensato non servissero... Luned� guardo su un libro che ho in ufficio e vedo se menzionano questa cosa. Conta che non ho mai aperto la scatola del laser di un confocale...
Citazione:
non ho capito l'astigmatismo dei fasci obliqui.
In pratica l'astigmatismo � un fenomeno per cui una lente mette a fuoco in modo diverso linee da direzioni diverse.
Tipico esempio � l'astigmatismo della cornea dell'occhio: chi soffre di astigmatismo ha la cornea curvata in modo diverso sull'asse orizzontale e quello verticale e quindi mette a fuoco diversamente le linee orizzontali da quelle verticali.
L'astigmatismo dei fasci obliqui � un fenomeno simile dovuto a imperfezioni della curvatura della lente. In questo caso l'astigmatismo si ha solo sugli oggetti fuori dall'asse principale della lente (la cui immagine arriva alla lente attraverso fasci di luce obliqui rispetto all'asse della lente). |
Sei un nuovo arrivato?
Leggi il regolamento del forum e presentati qui
My photo portfolio (now on G+!) |
|
|
|
carmilla983
Nuovo Arrivato
Prov.: al
Citt�: alessandria
59 Messaggi |
Inserito il - 24 febbraio 2008 : 10:02:54
|
ho l'esame domani quindi � urgenteeeee m aiuti a capire la unit scanning, please?
grasie |
|
|
|
gab
Nuovo Arrivato
1 Messaggi |
Inserito il - 05 marzo 2008 : 12:53:13
|
Salve!
Mi chiamo Gabriele.Vorrei sapere se qualcuno mi puo dare un parere riguardo a dei miglioramenti che, a mio avviso, potrebbero essere introdotti nella maggior parte dei microscopi confocali:
1) Sostituire uno degli "scanning mirrors"(la maggior parte delle pubblicazioni sono in inglese e penso che da noi si chiamino specchi a scansione..) con un deflettore ottico acustico allo scopo di accellerare la velocita' nel riprodurre le immagini finali (come unica controindicazione mi sembra di aver capito che questi deflettori siano decisamente costosi anche se non ho idea dei prezzi..)
2)Introdurre la tecinca del doppio fotone (La fluorescenza dovrebbe essere presente praticamente solo nel fuoco e non so quali possano essere le controindicazioni nell'usare tale tecnica..)
3)Sostituire, come rilevatori di segnale, i fotomoltiplicatori con dei fotodiodi, che mi sembrano essere piu' veloci con una migliore efficienza quantica (anche qui non vedo controindicazioni..)
Spero qualcuno parere a riguardo, sarebbe fantastico se mi si potesse dare qualche dritta su altri componenti che potrebbero essere sostituiti..
Ciao |
|
|
|
Discussione |
|



